




Increasing numbers of people are suffering from autism, a neurological developmental disorder. Those concerned experience problems with social interaction and communication. Cannabis could be an interesting treatment method. What anecdotal reports have been claiming for years, an Israeli study seems to confirm 2019. Yet many unanswered questions remain.
Autism or Autism Spectrum Disorders (ASD) are complex developmental disorders that affect the brain. There are various forms of the illness, where the person concerned demonstrates difficulties in the following areas:
- interaction with other people (social interaction)
- in their verbal and non-verbal communication
Repetitive and stereotypical behavioural patterns, interests and activities are also typical of the condition. Asperger’s Syndrome is a form of autism where cognitive development or language are in no way impaired.
Autism is much more common than many people think. According to the World Health Organization (WHO), 1 in 160 children suffer from ASD, whereas other sources name significantly higher figures. However, the dramatic rise in autism diagnoses worldwide is undisputed.

People assume that both genetic and environmental factors are aiding the development of autism, but a comprehensive explanatory model is yet to be established.
Cannabis as a treatment method for autism?
ASD is incurable, yet a number of treatment options exist that improve the lives of people with autism, including cognitive, psychological and pedagogical treatments.
Those patients who are being treated with cannabis are increasingly advocating its use. A few years ago, the famous author Marie Myung-Ok Lee spoke publicly about cannabis as a medicinal option for treating her autistic son:
“Marijuana isn’t a miracle cure for autism, but in our son’s case, it eases his pain and inflammation so dramatically that he is able to participate in life and learning again. It also protects him from the sometimes dangerous side-effects of pharmaceutical drugs. We have settled on a good strain (White Russian, a favourite form of pain-relief for end-stage cancer patients) and a good dosage. And now that he is no longer in pain, J. can go to school instead of a children’s psychiatric hospital, where all too many of his peers end up as a result of their violent behaviour.”
Associate Professor of Psychiatry, Emeritus at Harvard Medical School, Dr Lester Grinspoon, published a document on the use of cannabis in autism in 2010.
In this paper, Dr Grinspoon gives further detail into the experiences of Marie Myung-Ok Lee, who administers medicinal cannabis to her son J. The professor asks scientists to take these experiences seriously and to conduct further research into medicinal treatment:
“Anecdotal evidence commands much less attention than it once did, yet it is the source of much of our knowledge of synthetic medicines as well as plant derivatives. Controlled experiments were not needed to recognise the therapeutic potential of chloral hydrate, barbiturates, aspirin, curare, insulin or penicillin.”
In addition to the report by Marie Myung-Ok Lee, there are reports of many other children in whom autism symptoms have reduced after taking medicinal cannabis. Kalel Santiago, for example, is alleged to have spoken his first words after taking CBD. Just like with epilepsy, it seems that the majority of parents administer CBD oil to their autistic children, apparently with positive results. If activation of the cannabinoid receptors during the growth of a child is one of the causes of autistic disorders, it also needs to be considered if the administration of antagonists such as CBD could stop this effect.
In other cases reported in the media, the combination of THC and CBD proved to be more effective. Following these reports, those children suffering from both epilepsy and an autistic disorder are benefiting most from an increased THC content. There is also a case study concerning a six-year-old autistic boy whose symptoms have noticeably improved following treatment with the synthetic THC analogous compound dronabinol.
Alan Flashman, who has already treated over 500 autistic children with medicinal cannabis in Israel, comments on the optimal ratio of THC and CBD:
“The results are rather consistent. As I already reported, around 60% of children respond well to an oil with a ratio of 20:1 (CBD to THC). A further 15-20% require a higher THC content, often more THC than CBD. The last 20% continue to puzzle me somewhat; sometimes a change in cannabis variety makes a big difference. However, sometimes we have to acknowledge that the treatment has failed.”
The endocannabinoid system is fundamentally linked to autistic disorders
Over the past few years, some interesting research results concerning autistic disorders and the endocannabinoid system were published. It was also proven that the regions of the brain which have the highest concentration of CB1 receptors are those that we assume are dysfunctional in cases of autism, in particular the cerebellum, the hippocampus and the basal ganglia.
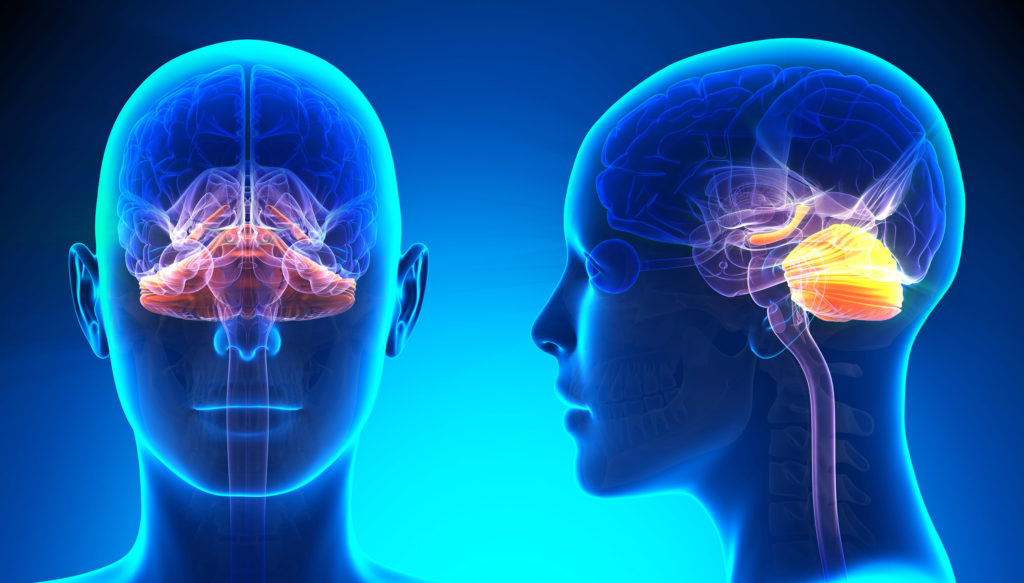
When the human foetus grows, the CB1 receptors and the associated endocannabinoids play an important role in differentiating between the neurons and the axonal migration; both processes are essential for normal neurological development. Other studies conclude that the CB1 receptors could be personally responsible for determining the synapse positioning. One theory is therefore that the activation of CB1 receptors in childhood could trigger autistic disorders because they interrupt the normal development of the brain.
The role of CB2 receptors in autistic disorders
CB2 receptors may also play a role in autistic disorders. It was proven that CB2 receptor agonists reduce the frequency with which certain important immune cells known as monocytes permeate the endothelium, i.e. the thin layer of cells separating the circulation from the tissues and organs. Monocytes are among the key cells associated with the immune system, and an interruption of their development and their functions was observed on multiple occasions in relation to the development of autistic disorders.
A study from the year 2013 showed that in children with autism, the concentration of CB2 receptors in the monocytes was increased, whereas the concentrations of CB1 receptors and the anandamide-reducing molecule Fatty Acid Amide Hydrolase (FAAH) were unchanged.
The potential role of the dopamine messenger system in autistic disorders
One case study reported on a middle-aged man who had previously been diagnosed with schizophrenia and who, due to psychotic symptoms, had sought acute psychiatric treatment, where he was first diagnosed with Asperger’s Syndrome. Schizophrenia and Asperger’s Syndrome have many common attributes and are therefore often confused.
Following treatment with the antipsychotic drug Aripiprazole, the Asperger’s symptoms improved considerably. Aripiprazole is a partial agonist on the dopamine D2 receptors, and authoritative evidence exists, showing that both schizophrenia and Asperger’s Syndrome are fundamentally linked to dopamine dysfunction.
It also looks as though anandamide plays a role in this process. It is already known that anandamide has a function for the dopamine messenger system, even if the precise mechanism cannot yet be properly understood. However, these studies indicate that one of the many functions of anandamide within the central nervous system could be to modulate psychomotor and social activities, which are primarily enabled through the dopamine D2 receptor.
Related post

What role do genes play in the development of autistic disorders?
Previously, it was assumed that the number of cases of autism attributable to genetic factors is 90%. Nowadays, it is assumed that the estimate was too high, because it was based on poorly invested studies of twins, and that the actual heritability of autism is only 50%.
One form of autism, the fragile X syndrome, is the most common monogenic (i.e. can be traced back to a mutation of an individual gene or chromosome) form of hereditary autism and is caused by an inactivation of the FMR1 gene, which is responsible for producing the protein FMR. It is known that the endocannabinoid system is involved in the control of cognitive functions such as anxiety, perception of pain and susceptibility to attacks, as well as the synaptic plasticity (the ability of the synapses to strengthen or weaken themselves depending on their activity level), and that all these characteristics are impaired in the case of Fragile X syndrome.
One study which investigated the role of the endocannabinoid system in male mice with a missing FMR1 gene through breeding discovered that when the CB1 receptors are blocked, cognitive damage, perception of pain and susceptibility to attacks normalised, whereas when the CB2 receptors were blocked, anxiety normalised.
Israeli study confirms that cannabis alleviates the symptoms of autism
As we have seen above, many patients advocate the use of cannabis for treating ASD symptoms. What is important to note here, however, is that these are individual findings which were not accompanied by strict empirical tests that ascertain that no other factors are responsible for the occurred effect.
These findings are insufficient for most doctors to recommend medicinal cannabis products containing THC to children, especially when you consider that there are also concerns regarding the effects of THC on brain development. What’s more, different manifestations of autism respond very differently to the administration of THC, and further research is required in order to determine more precisely what these responses could look like.
A 2019 Israeli study raises hope. Researchers at Ben-Gurion University of the Negev (BGU) and the Soroka University Medical Center confirm that cannabis represents a well-tolerated, safe, and effective form of treating ASD symptoms. It is still hoped that this will pave the way for future research. Double-blind, placebo-controlled studies are now particularly urgent, as requested by Dr Victor Novack of the BGU-Soroka Clinical Cannabis Research Institute.
- Disclaimer:This article is not a substitute for professional medical advice, diagnosis, or treatment. Always consult with your doctor or other licensed medical professional. Do not delay seeking medical advice or disregard medical advice due to something you have read on this website.


Loved reading this Post. Fair play to you. Have a 5 year old autistic son and this Post is definitely making me think about trying medical marijuana for his eating aversions. Good luck to you
After reading many articles on the effects of CBD oil on the endocannabinoid system I gave my 28 year old daughter CW oil.
She was diagnosed at 12 with depression, Bipolar, later anxiety, maybe Borderline Personality Disorder, possible PTSD and fibromyalgia w arthritis. A nurse mentioned she may have sensory processing issues when she was 24. Her psychiatrist recently diagnosed Aspergers. After one week on CW Every day Plus she stopped having episodes (seizures, sensory meltdowns, panic attacks). She slept longer and deeper. Needed much less pain med for muscle/joint pain. She was calmer, more peaceful, sweet. No angry, frustrated outbursts. She also was getting overheated which always added to anxiety and sensory issues. Less fatigue, happier, engaging more in conversation. Truthfully it’s all rather shocking! Charlottes Web CBD has made a huge impact on many lives.
Note: Not all CBD is effective.
I’m in New York and have no idea how to find a doctor (will travel) for my son who will consider cannibis treatment..any suggestions?
Get some Hemp based CBD which is currently legal at hemp stores or online.
You can find capsules or drops called tinctures. Start with 10 mg for a few days and see how he responds.
As a special needs teacher and marijuana advocate, one must be careful to assess each individual (and they themselves if cogent enough); the continuum suggests that some would benefit a lot, and some not so much.
That’s my gut feeling from working in “Ground Zero” type Education systems for a quarter century, from Protective custody in the prison system to students with standard scores under 70.
The bottom line is, more would benefit from cannabis than not I suspect. We just have to prove that for the naysayers, no small task indeed.