




Sickle cell disease (SCD) is a hereditary condition caused by a mutation in the haemoglobin gene, which if unmanaged can lead to symptoms of anaemia, extreme pain, and organ damage. People suffering from SCD are far more likely to use cannabis than the general population, potentially for its analgesic properties.
In sickle cell disease (also called sickle cell anaemia if there are two haemoglobin S genes), a mutation in the haemoglobin gene causes red blood cells to become distorted from the usual pliable, concave disc to a rigid, sickle-shaped form.
In normal conditions, the cells temporarily deform in order to be squeezed through narrow blood vessels. In SCD, the rigid cells are unable to pass, and can build up, blocking the blood vessels. This can lead to sickle cell ‘crisis’, an umbrella term for a set of acute conditions that may in extreme instances cause death within 24 hours. Loss of blood to the organs is one of the primary risks of sickle cell crisis, and usually causes cumulative damage leading to a shortened lifespan (although modern treatments have improved prognoses dramatically).
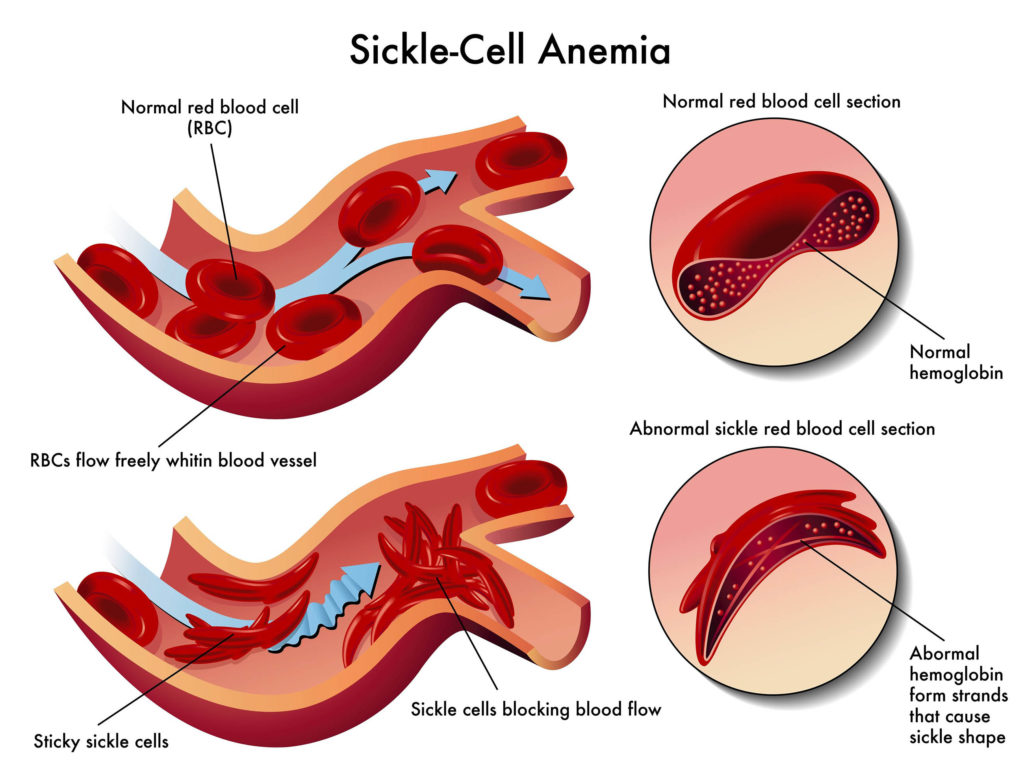
For someone to suffer from SCD, both alleles of the haemoglobin gene must be mutated; if just one allele is mutated and one is normal, the individual will exhibit both normal and sickled red blood cells and is considered a ‘carrier’.
The normal blood cells are able to produce over 50% of the requisite haemoglobin in carriers, and symptoms are not usually experienced unless triggered. ‘Triggers’ include lack of oxygen (e.g. at high altitude) and dehydration.
Sickle cell trait and malaria resistance
SCD is mainly found in (but is not exclusive to) tropical and subtropical populations, and those whose ancestry derives from such locations. It’s traditionally been thought that the sickle cell mutation occurred spontaneously in more than one location, and that positive selection pressures allowed it to take hold and flourish.
The possible reason for this is that possession of one mutated copy of the haemoglobin gene (also known as the sickle cell trait) is an advantage in areas high in malaria, as it confers a degree of resistance to the disease by mitigating the severity of its symptoms.
However, researchers have now discovered that this mutation can be tracked back to a child in Africa more than 7,000 years ago. This information might help doctors classify severity of cases and provide better care.
Inheritance and selection pressures
If two parents possess one mutated copy each, there is a 25% chance that the child will possess two mutated copies and will suffer from SCD. SCD sufferers don’t get the resistance to malaria. In fact, the pain associated with malaria is a major trigger for sickle cell crisis.
Although SCD is found in the descendants of tropical and subtropical populations now living in temperate zones such as Western Europe and much of the USA, the incidence is far lower than in populations still residing in their original locales. It’s thought that in areas where malaria isn’t endemic, possession of the sickle cell trait is not advantageous and is selected out over time.
Related post

Haemoglobin, oxygen and sickle cell disease
The mechanism of SCD is highly complex. A single-nucleotide polymorphism of the ß-globin gene causes the haemoglobin (the primary constituent of red blood cells) produced to contain valine instead of glutamic acid. Both of these substances are amino acids, but valine is hydrophobic, while glutamic acid is hydrophilic.
As blood plasma is mostly water, valine’s hydrophobic nature causes the mutated haemoglobin to collapse in on itself. Eventually, the haemoglobin begins to aggregate and polymerise into rigid, fibrous, sickle-shaped structures. This process occurs rapidly in times of low oxygen concentration, but is also promoted by high acidity and dehydration (reduced volume) of the blood.
Periods of low oxygen (hypoxia) in SCD patients are often followed by periods of reoxygenation. This is known as the hypoxia-to-reoxygenation cycle and is associated with severe pain (and possibly inflammation) at sites of haemoglobin aggregation (often areas rich in capillary blood vessels such as the lungs and genitals).
Haemoglobin is responsible for carrying oxygen to the organs of the body. In SCD sufferers, ischemia of the organs can result due to mutated blood cells building up and preventing healthy, oxygenated haemoglobin to pass.
How is sickle cell disease treated?
There are several ways to manage and control the various symptoms of SCD. The mainstay treatment is hydroxyurea, which reduces pain and complications from vaso-occlusive crises, reduces hospitalizations, and death from SCD. Infants born with SCD are given folic acid, which is continued into adulthood and often supplemented with penicillin in early childhood. In malarial countries, anti-malaria prophylaxis is often maintained for life, due to SCD sufferers’ increased susceptibility. However, a 2018 review in the efficacy of anti-malaria prophylaxis found no difference in adverse effects in children taking it versus children who were given a placebo.

In acute cases, blood transfusions can be performed, which increase the count of healthy red blood cells. The only known cure for SCD is bone marrow transplant, which is only known to be effective in children.
The only currently approved severe pain medications for SCD sufferers are opioids, many of which are dependence-forming and have significant side effects. The side effects often impact the kidneys (which are already at risk due to the disease itself) and can affect the blood vessels.
Sickle cell disease sufferers more likely to use cannabis
It’s been repeatedly observed that SCD sufferers use cannabis at higher rates than the general population, and sufferers have self-reported that cannabis use brings subjective feelings of pain relief. They also report it helps reduce anxiety and depression.
A 2018 study with 58 participants found that nearly half had used cannabis within the previous two years. The majority of them reported pain, anxiety, mood, sleep, and appetite as all being reasons for using, and 79% said it allowed them to reduce their use of pain medications.
In Jamaica (where cannabis is illegal, but use is widespread and socially accepted), SCD sufferers were found to use cannabis at almost twice the rate of the general population (women: 19% compared to 10%; men: 65% against 37%). This is despite the fact that there’s no expectation of ameliorated symptoms (only 6% of the sample associated cannabis use with management of symptoms).
The researchers noted that there appeared to be negligible difference in pain profile and number of pain events in their sample—however, the sample was small (just 145 people) and may not be indicative of trends on a larger scale.
Related post

Can medicinal cannabis help to cure sickle cell disease?
In 2010, researchers at the University of Minnesota found that the synthetic THC analogue CP 55940 was as effective as morphine sulphate in treating SCD-related severe pain in transgenic mice expressing human sickle haemoglobin. It was effective even at dramatically smaller doses than the opioid.
In 2011, a further paper submitted by the same researchers to Blood (the Journal of the American Association of Hematology) indicated that CP 55940 helped alleviate severe pain associated with the hypoxia-to-reoxygenation cycle. CP 55940 is a full agonist of both CB receptors, and is thought to act as an antagonist at the GPR55 receptor.
Cannabis has also been repeatedly shown to act as a vasodilator, which could in itself assist in easing the blockages caused by build-up of sickle cells. Conversely, there is some indication that cannabis may act as a vasoconstrictor in certain circumstances, particularly in the peripheral circulatory system (the capillaries), and that its use may even contribute to ischemic stroke (caused by obstruction of blood vessels leading to the brain). However, most studies drawing this conclusion investigated cannabis smokers who also used tobacco, a known vasoconstrictor.
Is medicinal cannabis preferable to opioids?
The answer is usually yes, if it can be shown to be as effective. The evidence for this isn’t bulletproof, but is definitely striking. Patients report a more positive side effect profile as a result of cannabis consumption compared to that experienced with opioids.

Sister Somayah Kambui, a medicinal cannabis patient and activist based in Los Angeles, California until her death in 2008, extolled the benefits of medicinal cannabis for SCD for many years—stating that morphine turned sufferers into ‘U.S. government junkies’ due to its hypnotic, soporific effects.
She had entirely replaced opioid use with that of cannabis and hemp oil. She managed her symptoms without incident, save for several occasions upon which her premises were raided and her cannabis confiscated. On one such occasion, a flare-up of symptoms resulted in hospitalisation and a return to the hated morphine.
Related post

More research is needed into cannabis and sickle cell disease
Sickle cell disease caused approximately 29,000 deaths worldwide in 2010 and it’s believed that at least two million people worldwide possess both copies of the mutated allele and are symptomatic.
Rates of SCD are on the increase. Using trend data from the World Health Organisation, authors of an article submitted to PLOS Medicine calculated that the number of babies born with SCD would increase to just over 400,000 by 2050 from just over 300,000 in 2010. Around 300 million people worldwide carry the sickle cell trait.
SCD is a painful and debilitating disease, and the overall inefficacy of opioid treatments and resultant poor quality of life for many sufferers is an indication that our approach to it is far from perfect. If cannabis is a good candidate to replace opioids, it should be implemented forthwith to prevent ongoing suffering for existing patients.
- Disclaimer:This article is not a substitute for professional medical advice, diagnosis, or treatment. Always consult with your doctor or other licensed medical professional. Do not delay seeking medical advice or disregard medical advice due to something you have read on this website.


My 8 year old has not long been diagnosed with SCD, I’m looking at treatment option, diet, vitamins. Minerals, natural therapy, pharma medications.
Its good to know that cannabis easy pain in scds compared to usual pain killers
But is there a therapy that cures scd for good
may just be a coincidence but the region with the highest prevalence of sickle cell disease is also where started the use of cannabis from the Bantu peoples with contacts with Indian zebu cattle breeders and Cannabis indica. Later Cannabis (diamba) was prohibited.